

-
製剤写真・
組成・性状 - お知らせ
- コード・番号表
- FAQ
- 電子添文
- インタビューフォーム
-
くすりのしおり
(日本語) -
くすりのしおり
(英語) - 適正使用ガイド
- RMP
-
患者向
医薬品ガイド - 製造販売後調査
- 患者指導箋
-
その他の
適正使用資料
製剤写真・組成・性状
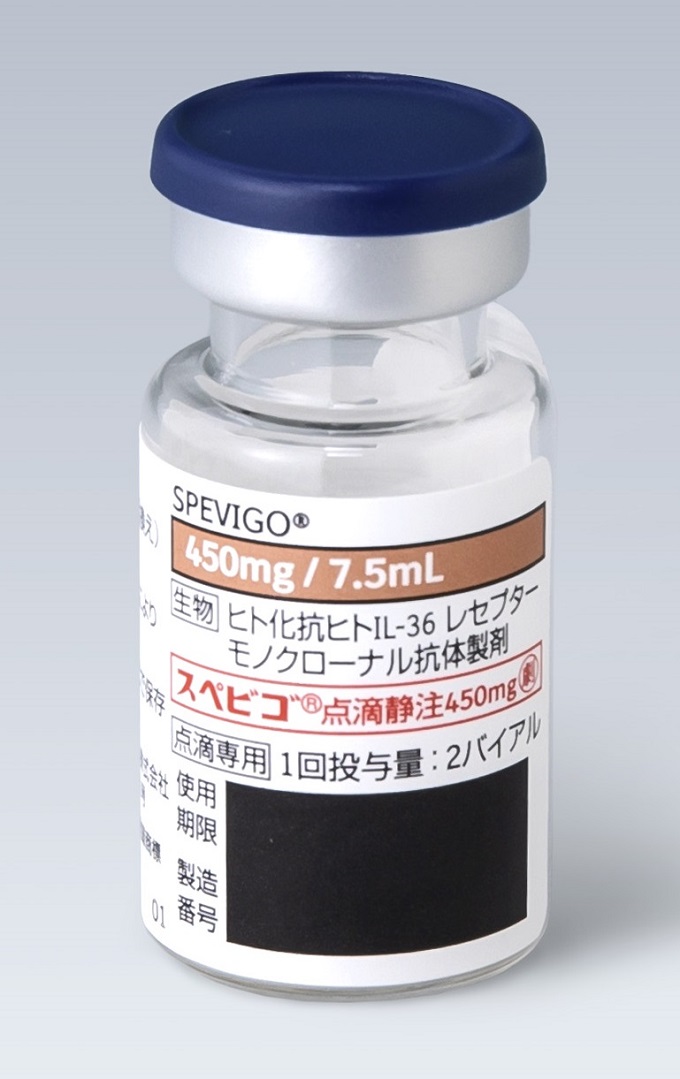
組成・性状
| 成分名 | スペソリマブ(遺伝子組換え) |
|---|---|
| 領域名 | 皮膚・炎症性・免疫性疾患 |
| 成分・含量 | 1バイアル7.5mL中 スペソリマブ(遺伝子組換え)450mg |
| 剤形・色調 | 注射剤(バイアル) |
| 性状 | 無色~微黄褐色の澄明又はわずかに乳白光を呈する液 |
| pH | 5.2~5.8 |
包装形態
コード・番号表
| 日本標準商品分類番号 | 873999 |
|---|---|
| 承認番号 | 30400AMX00409000 |
| 薬価基準収載 医薬品コード |
3999466A1026 |
| レセプト電算処理コード | 629920901 |
包装:7.5mL[2バイアル]
| JANコード | 4987413064118 |
|---|---|
| HOT7(処方用番号) | 1992090 |
| HOT番号 | 1992090010101 |
| 販売包装単位コード | (01)14987413064115 |
| 調剤包装単位コード | (01)04987413951135 |
適正使用ガイド
情報をシェアする