
-
製剤写真・
組成・性状 - お知らせ
- コード・番号表
- FAQ
- 電子添文
- インタビューフォーム
-
くすりのしおり
(日本語) -
くすりのしおり
(英語) - 適正使用ガイド
- RMP
-
患者向
医薬品ガイド - 製造販売後調査
- 患者指導箋
-
その他の
適正使用資料
製剤写真・組成・性状
組成・性状
| 成分名 | チオトロピウム臭化物水和物 |
|---|---|
| 領域名 | 呼吸器疾患 |
| 成分・含量 | 1カプセル中 チオトロピウム 18μg (チオトロピウム臭化物水和物として22.5μg) |
| 剤形・色調 | 明るい緑色の不透明の硬カプセル剤 |
| 内容物 | 白色の粉末 |
| 外形 |
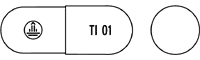
3号 |
| 識別コード | 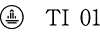 |
| 長さ | 約16mm |
| 直径 | 約6mm |
| 重さ | 約0.054g |
剤形

包装形態


吸入用器具(ハンディヘラー®)

吸入用器具(ハンディヘラー®)
コード・番号表
| 日本標準商品分類番号 | 872259 |
|---|---|
| 承認番号 | 21600AMY00131000 |
| 薬価基準収載 医薬品コード |
2259709G1027 |
| レセプト電算処理コード | 620002421 |
包装:14カプセル
(ハンディヘラー付)
| JANコード | 4987413826037 |
|---|---|
| HOT7(処方用番号) | 1165791 |
| HOT番号 | 1165791010101 |
| 販売包装単位コード | (01)14987413826034 |
| 調剤包装単位コード | (01)04987413950589 |
包装:28カプセル
(ハンディヘラー付)
| JANコード | 4987413826051 |
|---|---|
| HOT7(処方用番号) | 1165791 |
| HOT番号 | 1165791010201 |
| 販売包装単位コード | (01)14987413826058 |
| 調剤包装単位コード | (01)04987413950589 |
包装:28カプセル
| JANコード | 4987413826129 |
|---|---|
| HOT7(処方用番号) | 1165791 |
| HOT番号 | 1165791010102 |
| 販売包装単位コード | (01)14987413826126 |
| 調剤包装単位コード | (01)04987413950589 |
よくある質問
-
吸入用器具ハンディヘラーは金属およびプラスチックからできております。患者様が廃棄の際には、地方自治体の廃棄方法に従うようご指導ください。 ハンディヘラー内部には、カプセルに穴を開けるための針があり、危ないので分解しないようにしてください。
-
チオトロピウムは腎排泄型であり、腎機能低下患者様では血中濃度の上昇がみられるため、腎機能が高度あるいは中等度低下している患者様(クレアチニンクリアランス値が50mL/min以下の患者様)には慎重に投与してください。
腎機能が低下している高齢者に対して本剤を投与する場合には、治療上の有益性と危険性を勘案して慎重に投与し、有害事象の発現に注意してください。<参考>
腎機能低下患者(海外)においては、チオトロピウムの静脈内投与及び吸入投与後の血漿中未変化体濃度は上昇し、腎クリアランスは低下した。軽度の腎機能低下患者(クレアチニンクリアランスが50~80mL/minの患者、海外)において、チオトロピウム4.8μgを静脈内投与後のAUC0-4hは健康成人(海外)に比較して39%高い値を示した。また、高度あるいは中等度の腎機能低下患者(クレアチニンクリアランスが50mL/min未満の患者、海外)においては血漿中未変化体濃度は約2倍高い値を示した(AUC0-4hは82%高かった)。<引用>
スピリーバ吸入用カプセル 電子添文 -
一般に高齢者では腎クリアランス等の生理機能が低下しており、血中濃度が上昇するおそれあるので、副作用の発現に注意してください。臨床試験で口渇は高齢者でより高い発現率が認められています。
腎機能が低下している高齢者に対して本剤を投与する場合には、治療上の有益性と危険性を勘案して慎重に投与し、有害事象の発現に注意してください。<参考>
高齢者(海外)では、チオトロピウム18μgを吸入投与後の腎クリアランスは低下した(腎クリアランスは58歳以下の慢性閉塞性肺疾患患者で326mL/min、69歳以上の慢性閉塞性肺疾患患者で163mL/min)が、これは加齢に伴う腎機能の低下によるものと考えられた。
若年健康成人(平均年齢32.1歳、海外)にチオトロピウム108μgを吸入投与したときの尿中未変化体排泄率は14%であったが、慢性閉塞性肺疾患患者(平均年齢63.8歳、海外)にチオトロピウム18μgを吸入投与したときの尿中未変化体排泄率は7%であり、若年健康成人に比較して低い値であった。
一方、高齢者(海外)にチオトロピウム18μgを1日1回反復吸入投与後のAUC0-4hは非高齢者(海外)に比較して43%高い値を示した。非高齢者及び高齢者における薬物動態パラメータは以下のとおりであり、個体間変動を考慮すると、血漿中未変化体濃度に加齢による大きな差はないと考えられた。<引用>
スピリーバ吸入用カプセル 電子添文 -
本剤の小児等(低出生体重児、新生児、乳児、幼児又は小児)を対象とした試験は実施しておらず、安全性は確立していないことから、小児への使用は承認されていません。
<引用>
スピリーバ吸入用カプセル 電子添文
スピリーバ吸入用カプセル インタビューフォーム Ⅷ.安全性(使用上の注意等)に関する項目 11.小児等への投与 -
本剤の妊婦に対する臨床試験成績はなく、安全性は確立していないことから、妊婦又は妊娠している可能性のある婦人には、治療上の有益性が危険性を上回ると判断される場合にのみ投与してください。
<参考>
動物実験(ラット)で胎児に移行することが認められている。<引用>
スピリーバ吸入用カプセル 電子添文
スピリーバ吸入用カプセル インタビューフォーム Ⅷ.安全性(使用上の注意等)に関する項目 10.妊婦,産婦,授乳婦等への投与
